ABSTRACT
An increasing body of evidence suggests that RNA- and ribonucleoprotein-based regulation plays a significantly more central role in controlling gene expression than previously thought. This regulation may extend as microparticle-based intercellular exchange of RNA and associated RNA-binding proteins, whereby cells are able to influence each other’s gene expression mechanisms at multiple levels and thus coordinate phenotypes. My data suggests that the 3’untranslated region of Tissue Factor and of CXCL10 is sufficient to trigger shuttling of an associated 5’GFP mRNA between physically isolated HeLa cells, such that recipient cells express GFP. By contrast, GFP mRNA absent the required UTR does not shuttle. Analysis of the culture supernatant reveals that GFP/3’UTR mRNA is present in a fraction commonly associated with microparticles. These results suggest the existence of a mechanism by which thrombogenic and inflammatory responses could be coordinated between cells by microparticle-based mRNA exchange.
ABBREVIATIONS
ARE: AU-rich element
BMDM: bone marrow-derived macrophage
BSA: bovine serum albumin
CMV: cytomegalovirus
CXCL10: C-X-C motif chemokine 10
DMEM: Dulbecco’s modified Eagle’s medium
DNA: deoxyribonucleic acid
dNTP: deoxyribose nucletode triphosphate
eIF3: eukaryotic initiation factor 3
FCS: fetal calf serum
GFP: Green Fluorescent Protein
HuR: Human antigen R
HUVEC: Human umbilical vein endothelial cell
IFNg: Interferon gamma
miRNA: micro-RNA
mRNA: messenger RNA
ORF: open reading frame
PARP14: Poly (ADP-ribose) polymerase 14
PBS: phosphate buffered saline
PCR: polymerase chain reaction
RBP: RNA binding protein
RNA: ribonucleic acid
RNP: ribonuceloprotein
RT-PCR: reverse transcription PCR (non-quantitative)
TF: Tissue Factor
TTP: tristetraprolin
USER: untranslated sequence element for regulation
WB: western blot
INTRODUCTION
The perception of eukaryotic gene expression has been transformed over the past decade as the extent and complexity of post-transcriptional regulation has shifted into focus. Traditionally, gene expression has been thought principally determined by the combinatorial influence of repressive or activating transcription factors on promoter sequences within the context of local chromatin structure [1]. Yet transcription is of a spontaneous, stochastic nature and is prone to cross-activation between spatially adjacent genes. The impact of post-transcriptional regulation is to significantly augment regulatory stringency; the physiological import of which is highlighted by the abundance of RBPs, which in eukaryotes outnumber DNA-binding proteins nearly two-to-one [2]. Indeed the correlation between steady state mRNA and protein levels has been reported weak to moderate at best [2]. Compellingly, the breadth and width of RNA-orientated regulation are consistent with the notion of the RNA World, wherein protein biosynthesis predated DNA and was wholly dependent on RNA [1].
mRNA fate may be influenced at every step from transcription to translation. Within the nucleus, trans-acting RBPs quickly associate with nascent mRNA and regulate alternative splicing and polyadenylation, RNA editing, and post-transcriptional modifications to allow a diversity of variants to arise from relatively few protein-coding genes. Splicing, furthermore, allows RBPs to mark transcripts in such a way as to guide future regulatory steps, including translation [2]. Mature transcripts are then exported from the nucleus to the cytoplasm [1], where miRNAs, and RBPs regulate the stability, localization, and translation of functionally-related transcripts in a coordinated manner to form regulatory modules are known as “RNA operons” [1].
The constituent regulatory factors of RNA operons interact with several sequence elements within mRNA known as USERs [1]. USERs may allow cooperation or competition of regulatory factors. In turn, these regulatory factors may be multi-targeted and may belong to different RNA operons [1]. Moreover, mRNA coding for a specific protein which functions in multiple biological contexts may form part of different operons and thus be differentially regulated [1]. This higher order complexity can be conceptualized as “RNA regulons” [1], which form the basis of a cellular RNP-based regulatory system that acts at multiple levels of gene expression, and which is able to both feedforward onto the proteome as well as feedback onto early RNA processing machinery [2].
Over recent years, a new phenomenon has emerged which may reveal that this RNP-based regulatory system is not simply confined to the cellular level, but rather is able to orchestrate gene expression on the intercellular stage. mRNAs, miRNAs, and RBPs have been discovered included amongst material cargo shuttled between cells via membrane-bound bodies dubbed “microparticles” [3]. Microparticles refer to a heterogeneous population whose members include endosome-originated “exosomes” (30-120 nm in diameter) and vesicles shed directly from the cellular membrane, known as “microvesicles” (0.1-1 mm in diameter) [4]. This microparticle shuttle phenomenon is but one of multiple methods by which cells interact in order to coordinate their behaviour within tissues; other methods include the exchange of soluble factors, physical interaction through cell adhesion molecules, cytonemes allowing the exchange of membrane bound factors, and tunneling nanotubules that in addition allow the exchange of cytoplasmic components [5].
Microparticle shuttle differentiates itself from these other forms of communication in that the particle membrane encapsulates a microenvironment protected from the vicissitudes of the extracellular fluid; yet by virtue of possessing an independent membrane, the microparticle is able to travel long distances from its originating cell. Microparticles are found in abundance in biological fluids and have varying composition depending on the cell of origin [4]. By virtue of microparticle-cell surface interactions, long-range communication by microparticle shuttle would be able to target very specific sub-populations of cells [4]. Microparticles thus exhibit a number of traits that make them highly suitable as carriers of nucleic acids and RNP complexes. Potentially, therefore, microparticles could shuttle RNP complexes or even entire RNA operons between cells that act on multiple levels of gene expression, thereby altering recipient cell phenotype [1]. Such a phenomenon would have important implications for development and pathogenesis in eukaryotes.
My interest in this area stems from some of my earlier work with an IFNg-inducible RBP known as PARP14. My pilot data suggested that PARP14/GFP- fusion protein was transferred between HeLa cells via tunneling nanotubules (Figure 1), potentially illustrating an intercellular transfer mechanism for RNP complexes.

PARP14 regulates mRNA stability; notable examples include TF and CXCL10, with whose 3’UTRs PARP14 directly interacts [6]. CXCL10 is an IFNg-inducible chemokine involved in inflammatory responses and leukocyte trafficking; whilst TF is a membrane-bound glycoprotein that binds factor VIIa to initiate pro-coagulatory signaling, especially in disease contexts [7]. In macrophages, CXCL10 mRNA- and TF mRNA-bound PARP14 has been shown to further complex with HuR or TTP, which are themselves RBPs that also regulate mRNA stability [6]. In general, HuR and TTP share numerous mRNA targets, including cytokines and chemokines that require coordinate regulation during immune responses [1].
Suggestively, it has already been reported that HuR is present in Jurkat T-cell-derived exosomes and that in these cells, HuR associates with a pool of mRNA targets that share extensive overlap with mRNA present in mast cell-derived exosomes [2]. Consequently, HuR has been proposed as a regulator for the intercellular exchange of RNP complexes. Conceivably therefore, HuR and/or PARP14 complexes may be involved in the shuttling of TF and/or CXCL10 mRNA. If true, this would be of great import to immunologic regulation.
A concrete immunological role has already been established for microparticle shuttle. Indeed the discovery that microparticles participate in antigen presentation was the initial evidence that these membrane fragments are not simply the result of cellular damage, as was initially thought [5]. Circulating microparticles are known to be elevated in many diseases and to play a part in inflammatory responses and thrombogenesis [8]. For example, exposure to TF protein-bearing microparticles has been shown to increase the thrombogenicity of endothelial cells, including their own expression of TF8. Macrophage- and platelet-derived microparticles have been reported at the core of artherosclerotic plaques, where they are thought to be involved in inflammatory and prothrombotic signaling [5]. Platelet-derived microparticles have been shown to promote haemotopoetic cell survival as well as the binding of monocytes to the vascular endothelium [9]. CXCL10 protein has also been reported in microparticles [3], although its function has not been investigated.
In summary, PARP14 and HuR are both RBPs involved in regulating mRNA stability. They both appear to be trafficked between cells. They interact with each other (intracellularly). PARP14 regulates TF and CXCL10 mRNA. TF and CXCL10 protein have been shown to leave the cell via microparticle shuttle. Microparticles have been shown to be immunologically involved and to traffick mRNA as well as protein. Accordingly, my hypothesis is that TF and/or CXCL10 mRNA are trafficked between cells via microparticle shuttle, possibly in complex with PARP14 and/or HuR.
The data below suggests that this hypothesis is correct, although they are not conclusive. Using chimeric genes consisting of 5’GFP and the 3’UTR of either TF or CXCL10 in a plasmid vector, I established that donor cells transfected with construct DNA release construct mRNA into the culture medium. Sterile transfer of this medium to untransfected recipient cells results in the intracellular presence of both construct mRNA and GFP protein. This transfer between donor and recipient cells is dependent upon the 3’UTR of the constructs and does not occur with a GFP-only plasmid. Furthermore, the intracellular and cell population distribution of GFP protein in recipient cells after shuttling is completely unlike what is seen in cells directly transfected with GFP-only plasmid DNA. Finally, I demonstrate that 3’UTR-containing mRNA is present in a fraction of culture supernatant normally associated with microparticles.
RESULTS
With a view towards simplifying the technical requirements of the research, chimeric genes were engineered consisting of a 5’GFP ORF conjugated to the murine 3’UTR of either TF or CXCL10. The inclusion of only the 3’UTR focused this project on a single regulatory region; one that is commonly associated with the regulation of mRNA stability and that directly interacts with PARP14, which pilot work has suggested is trafficked from cells. The expectation was that if either TF or CXCL10 mRNA is shuttled between cells in vivo, the relevant 3’UTR might be sufficient to trigger the mechanism involved. The use of murine sequences were intended to facilitate possible future experimentation in mouse BMDMs. No compatibility issues were expected for their use in HeLa cells due to the evolutionary conservation between murine and human TF and CXCL1011 as well as the highly generic nature of mammalian RBPs11. The GFP moiety served as an easily differentiable marker, being not present in normal HeLa cell culture.
These constructs were engineered as described in Methods. Briefly, the relevant 3’UTR sequence was amplified through PCR from already available macrophage lysate. Gel electrophoresis verified the length of the resulting DNA sequences (Figure 2A,B). This DNA was purified and TOPO cloned into a commercial plasmid vector containing the aforementioned 5’GFP under the control of a CMV promoter. This plasmid was then transformed into competent E. coli, which were plated and grown. For each construct, 8 colonies were picked and separately grown in broth. From this broth, plasmid DNA was purified and sent for sequencing using orientation-specific plasmid primers. Sequencing traces were clear and analysed with BioEdit (Figure 2C,D). One clone for each construct was chosen with no sequence mutations and regrown in broth to yield endotoxin-free construct DNA.

HeLa cells were transiently transfected with either GFP/TF-3’UTR or GFP/CXCL10-3’UTR DNA as described in Methods, or subjected to the transfection protocol absent DNA as a control, and incubated for 16 hours at 37oC. These cells were then lysed and RNA purified from the lysates. RT-PCR yielded cDNA in a construct specific manner by using a forward primer specific for GFP and a reverse primer specific for either TF or CXCL10. Gel electrophoresis of this cDNA (Figure 2E,F) revealed that construct mRNA was present in the cells, indicating successful transfection and expression. WB of similarly prepared cell lysates demonstrated that the GFP/TF-3’UTR construct was successfully expressed as GFP protein (Figure 2G), however time constraints precluded a similar check being performed for GFP/CXCL10-3’UTR.
mRNA shuttle phenomenon (preliminary)
A simple preliminary assay was performed to determine whether transfer of culture supernatant from construct-transfected HeLa cells to untransfected HeLa cells would result in transfer of mRNA and subsequently GFP expression. If the 3’UTR in the construct mRNA is indeed sufficient to stimulate intercellular mRNA shuttle, then such shuttling should occur through supernatant transfer, barring cytoneme- or nanotubule-based mechanisms.
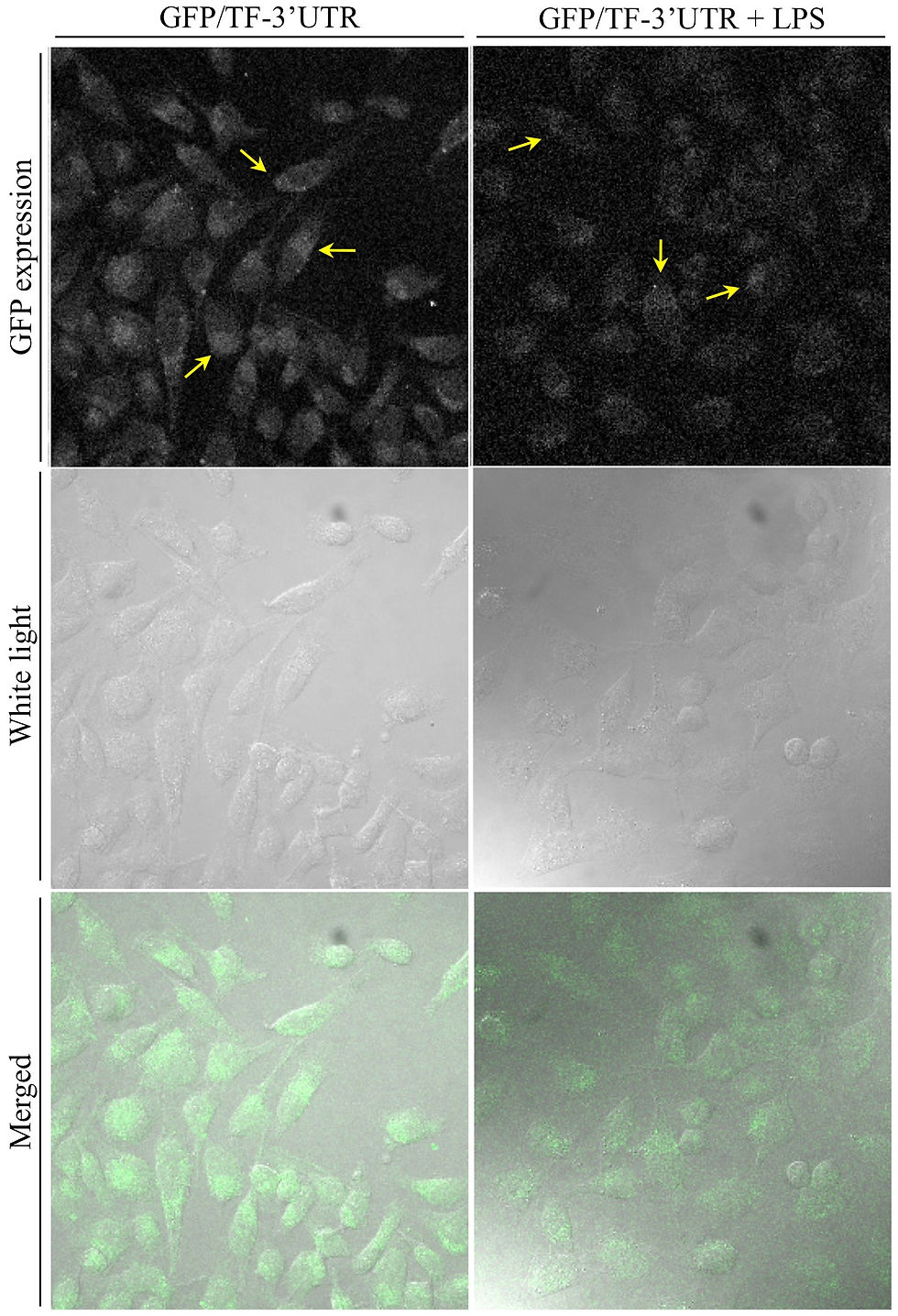
In order to address this, HeLa cells were transiently transfected with either GFP/TF-3’UTR or GFP/CXCL10-3’UTR DNA, or subjected to the transfection protocol absent DNA as a control. These cells were incubated for 16 hours at 37oC. GFP/TF-3’UTR-transfected cells and GFP/CXCL10-3’UTR-transfected cells were then either stimulated with LPS (1 mg/ml) or IFNg (2.5 ng/ml) respectively, or left unstimulated. These cells were then incubated for a further four hours at 37oC, after which the cell culture medium was collected and centrifuged at 1000g for 5 min to pellet cells and debris. The supernatant was transferred to medium-free, untransfected Hela cells, which were then incubated for 16 hours at 37oC. Finally, the untransfected cells were washed of culture medium with PBS (twice), then either lysed and assayed for construct mRNA using RT-PCR as described previously, or fixed and mounted (unstained) for confocal microscopy as described in Methods.

In the cases of both constructs, the untransfected (recipient) cells having received culture supernatant from transfected (donor) cells expressed construct mRNA regardless of donor cell stimulation (Figure 3), and also expressed GFP protein (Figures 4,5). As expected, the control recipient cells expressed neither construct mRNA nor GFP protein (Figures 3,6). The donor cell lysates (positive control) were accidentally disposed of and so are not present in Figure 3. Although encouraging, these preliminary results only demonstrate that either mRNA or plasmid DNA was transferred between cells, either free in the culture supernatant or encapsulated in some lipid vehicle.
This transfer of nucleic acids suggests that the GFP protein observed in recipient cells was not simply transferred via supernatant, subsequently becoming adherent to cell surfaces. Moreover, the recipient cells exhibited punctuate GFP distribution as well as larger localized regions of fluorescence (Figure 4,5). Intriguingly, the entire recipient cell populations expressed GFP protein: a contrast to directly transfected cells, in which only a subpopulation of the cells in the well become transfected even under the best of circumstances. This argues against plasmid DNA being the causative agent.


mRNA shuttle phenomenon (further investigation)
In order to address the aforementioned issues regarding the potential mRNA shuttling being observed, the culture supernatant transfer experiment was repeated as previously described with a number of modifications. Firstly, the supernatant obtained after centrifugation of the culture medium was treated with various enzymes, the combined results of which could be used to gain insight into the underlying cause of the observed phenomenon. Some supernatant was treated with DNase I, to degrade plasmid DNA in the supernatant leftover from transfection of the donor cells; with RNase A, to degrade mRNA in the supernatant; and with a combination of Proteinase K and RNase A, the Proteinase K to possibly disrupt proteins on the surface of microparticles so as to inhibit microparticle recognition by recipient cells and/or destabilize microparticle structure to render internal mRNA susceptible to degradation by RNase A. The DNase I was removed before transfer to recipient cells, however resources were not available to remove RNase A or Proteinase K. Secondly, a UV control was used to compare exosome-producing cells with apoptotic cells12. 4 hours after transfection a separate source of donor cells were washed of culture medium with PBS twice, given fresh medium and irradiated with UV light for 10 min at 180 W. These cells were replaced in the incubator for a further 16 hours. The morphology of the cells was observed by light microscopy then the culture medium was collected as normal. Thirdly and most importantly, the control donor cells were transfected with GFP-only (including polyA-tail but absent 3’UTR) plasmid DNA rather than no DNA. In addition to these modifications, LPS and IFNg stimulation were omitted, as they appeared to have no significant effect.
This repeat of the supernatant transfer experiment was unsuccessful in all instances for GFP/CXCL10-3’UTR for reasons unknown (Figure 7A), although the donor cell lysate (positive control) reveals successful transfection. For GFP/TF-3’UTR, recipient cells expressed construct mRNA in all cases except when receiving from UV-irradiated donor cells (Figure 7B). The morphology of these UV-irradiated donor cells was verified by light microscopy (not shown) prior to collection of the culture medium. The irradiated cells appeared spherical yet still adhered to the well surface, discounting the possibility of necrosis and the accompanying release of DNases and RNases into the culture medium. Therefore it appears shuttling does not occur from apoptotic to healthy HeLa cells. The remaining recipient cells yielded cDNA bands of different intensities, although the reliability of such differences in non-quantitative RT-PCR is suspect. RNase A-treated supernatant induced a more intense band than for untreated supernatant. By contrast RNase A/Proteinase K treated and especially Dnase I treated supernatant induced fainter bands than untreated supernatant.

A control experiment was conducted in which 2 mg of TF construct DNA was spiked directly into 2ml of sterile medium and treated with DNase I in parallel with the culture supernatant. This same quantity of DNA is used for transfection and is within the manufacterer’s advertised processing capability of DNase I. Nevertheless, PCR amplification revealed that TF construct DNA was still present in the sterile medium (Figure 7C). DNAse I was therefore non-functional in the context of the culture medium. A viable cell count was made of the recipient cells prior to lysis and RNA purification, using Trypan blue. The viable cell concentration for cells receiving DNase I-treated supernatant was 1.67 x 104 cells/ml as opposed to 7.67 x 104 cells/ml (average of 3 readings) for untreated supernatant, although the statistical significance of this cannot be determined as n=1. Other types of recipient cells have similar viable counts to those that received untreated supernatant. Consequently, the fainter band associated with DNase I in Figure 7B is likely due to toxicity of the buffer involved, which could not be removed prior to transfer.
No buffer was used in the RNase A and RNase A/Proteinase K treatment. The intensity of the associated bands is subject to speculation. A cellular response to the presence of the enzymes may be involved somehow. The GFP plasmid control did not exhibit shuttling of GFP-only mRNA (Figure 7D). Hence the shuttling process is not the result of random plasmid transfer, as evinced by its specificity for the 3’UTR of TF.
In a parallel experiment, culture supernatant from HeLa cells transfected with GFP/TF-3’UTR DNA was either treated with DNase I or left untreated and transferred to untransfected cells as previously described. The control again involved GFP-only plasmid DNA rather than no DNA. After the 16 hour incubation at 37oC with donor cell supernatant, the recipient cells were treated with the nuclear stain TOPRO and mounted for confocal microscopy. The donor control cells were similarly prepared. Regardless of DNase I treatment (now known to be non-functional under experimental conditions), recipient cells demonstrated GFP distribution (Figure 8) similar to that observed previously. Importantly, recipient control cells exhibited no significant fluorescence, whilst donor control cells that were directly transfected with GFP plasmid exhibited a diffuse pattern of florescence (Figure 9) completely unlike the product of mRNA shuttle.
This difference in fluorescence patterns could possibly be caused by cellular recognition of the 3’UTR or of the exogenous nature of shuttled mRNA that triggers funneling down a specific processing route within recipient cells. As previously mentioned, only a subpopulation of donor control cells were transfected, as is common in direct transfection and in contrast to mRNA shuttle, where apparently all cells exhibit fluorescence. This provides further evidence for the non-plasmid nature of the transfer phenomenon observed. Finally, a Z-stack series of micrographs were taken of sample recipient cells. Sample images from this series (Figure 10) demonstrate that the GFP protein is truly intracellular and not adhered to the surface of the cells.



Fractionation of culture medium
Having established that a phenomenon of shuttling occurs between transfected and untransfected HeLa cells, this project investigated the vehicle by which this shuttling occurs. HeLa cells were transfected with GFP/TF-3’UTR DNA, or with no DNA as a control. The cells were incubated for 16 hours at 37oC, after which the cell culture medium was collected and differentially centrifuged at 5 000g for 10 min, at 10 000g for 10 min, at 18 000g for 30 min, and at 22 000g for 30 min. RNA was purified from each of the four resulting pellets, as well as from the leftover supernatant and the original cell lysates (positive control). cDNA was prepared as previously described. GFP/TF-3’UTR mRNA was present in all fractions except for the 22 000g pellet (Figure 11).

This experiment was repeated for both GFP/TF-3’UTR and GFP/CXCL10-3’UTR constructs. This time the culture medium was centrifuged at 5 000g for 10 min, 22 000g for 10 min, and 150 000g for 60 min. Construct mRNA was present in all fractions in this case (Figure 12A,B). However, the supernatant fraction contained almost no mRNA, whilst a significant amount was present in the 150 000 g fraction, which is commonly accepted to pellet microparticles.

DISCUSSION
To recapitulate the previous section, plasmid vectors were engineered that encode chimeric genes for 5’GFP conjugated to the murine 3’UTR of either TF or CXCL10. These constructs were then transfected into HeLa cells and various assays performed, from which four principle findings ensued. First, the transfer of culture supernatant from transfected to untransfected cells results in the transfer of both construct mRNA and GFP protein expression. Second, the intracellular and cell population distribution of GFP protein in such recipient cells is completely unlike what is seen in cells directly transfected with plasmid DNA encoding only a GFP gene and its associated polyA-tail. In recipient cells, GFP exhibits punctuate distribution as well as larger regions of localized concentration. By contrast, directly transfected cells have a diffuse pattern of GFP distribution throughout the cell. Furthermore, the recipient cell population homogeneously expresses GFP, whilst in directly transfected cells only a subpopulation of cells express GFP. Third, a comparison of GFP/TF-3’UTR plasmid with the GFP plasmid revealed that GFP/TF-3’UTR mRNA exhibits shuttling (via supernatant transfer) whilst GFP mRNA does not. Shuttling is therefore dependent on the presence of specific 3’UTR sequences. Fourth, GFP/TF-3’UTR and GFP/CXCL10-3’UTR mRNA were isolated in a pellet from centrifugation at 150 000 g after all cell debris and apoptotic bodies had been cleared by lower speed centrifugation. This pellet is widely accepted to contain microparticles (likely both exosomes and microvesicles) [4]. Supporting evidence is thus provided for the particulate nature of the vehicle mediating mRNA shuttle, as was initially postulated.
Further data include the absence of shuttling from UV-irradiated donor cells and the presence of shuttling despite various enzymatic treatments of donor cell supernatant. The UV-control was originally based on the fact that apoptotic cells secrete apoptotic blebs but not exosomes [12] and thus was intended to simultaneously provide evidence for both an active cellular process and an exosome-based vehicle. However the use UV light may also have caused strand breakage and/or crosslinking of mRNA and plasmid DNA [13]. The UV control therefore does not disprove plasmid-based transfer (in membrane compartments or free in supernatant) or mRNA-based transfer (in apoptotic bodies or free in supernatant). The possibility of nucleic acid degradation unfortunately renders this control uninformative.
With regards to enzymatic treatment of the transferred supernatant, there were too many variables that were not controlled for the results to be informative. Although DNase I was removed prior to transfer, RNase A and Proteinase K was not. Prolonged exposure to these enzymes by the recipient cells may have affected cell viability and/or triggered cellular responses that may have affected cellular inclination towards receiving shuttled mRNA. A further complication is that 1ul of Proteinase K is able to degrade RNase A, although previous studies indicate that in 60 min, the 100 ul of RNase A will have had time to act before being degraded [14]. It is tempting to suggest that the relative intensities of the RNase A- and RNase A/Proteinase K-associated bands are such because, as predicted, the shuttled mRNA was encapsulated within microparticles that offered protection against RNases but whose structural integrity was protein-dependent and so was compromised by proteases. Yet this remains pure speculation, especially as the viable cell count was statistically unreliable (n=1). Certainly the observation of clumps of floating cells as well as the lower viable cell count for recipient cells having received DNase buffer strongly suggests that the lower intensity of the associated cDNA band is due to less cell material. The observation that DNase I was non-functional in the experimental context supports this conclusion.
UV irradiation and enzymatic treatment would therefore require a complex experimental setup in order to yield informative results. In retrospect, a simpler assay would have been alkalinisation of the transferred supernatant to trigger hydrolysis of free mRNA. Additionally, the use of an alkaline-tolerant phospholipase would have been more appropriate than Proteinase K for the disruption of microparticle integrity and could have been used to render phospholipid-encased mRNA susceptible to alkaline hydrolysis. Nevertheless, the results presented herein are suggestive that mRNA shuttling (rather than protein or DNA transfer) occurs between transfected and non-transfected cells. Furthermore, it appears that this process is dependent on the 3’UTR of TF and of CXCL10, and that the shuttled mRNA is translationally functional. This same mRNA is also present in a fraction of culture medium commonly associated with microparticles, however no data links this specific mRNA to the shuttle phenomenon nor provides direct evidence of the presence of microparticles in this fraction. Alternatively therefore, the observed transfer phenomenon may be due to the transfer of free mRNA in the supernatant that is directly taken up by recipient cells. Speculatively, this could occur “accidentally” through constitutive pinocytosis. The mRNA would then presumably be recognized somehow via its 3’UTR and escape lysosomal/endosomal degradation.
Regardless, in the short-term, demonstrations of the shuttling of TF-3’UTR- and of CXCL10-3’UTR-containing mRNA would have to be repeated for statistical significance. This should be contrasted with repeated demonstrations of the absence of shuttling of control mRNA that contain scrambled TF-3’UTR- and CXCL10-3’UTR sequences. In order to investigate the postulated microparticle vehicle responsible for shuttling, sucrose flotation could be used [3]. Here, the exosome pellet is resuspended in 2.5 M sucrose, on top of which a linear gradient of 2.0 M-0.25 M sucrose is layered. The sucrose gradient is then centrifuged at high speed, during which the low-density particles, such as exosomes and microvesicles, float to a similar density of sucrose. Different fractions can then be assayed for DNA, RNA, and protein content. Exosomes have a characteristic density in the range of 1.11-1.21 g/ml^3. The colocalisation of 3’UTR mRNA with CD63 (a common surface marker of exosomes [3]) at this sucrose density would provide evidence for the exosomal nature of the observed shuttling. Furthermore, sucrose flotation is an effective method to purify exosomes [3], which could then be utilized for further experiments, such as RNase treatment to determine whether the mRNA is external or internal to the microparticles. Additionally, purified exosomes could be irradiated with UV light to crosslink RNA and adjacent proteins. In this way the identity and position of RBPs associated with the 3’UTR mRNA would be revealed [15].
In the medium term, experimentation would progress to the use of full-length TF or CXCL10 to replace GFP. The continued use of murine sequences would enable the identification of construct mRNA and protein in human cells. The supernatant transfer experiments could be repeated using transfected macrophages as donor cells and HUVECs16 as recipient cells, as this would mirror the nature of the cells in vivo where the physiological equivalent of this shuttling phenomenon is hopefully occurring: i.e. between stimulated macrophages and the surrounding endothelial cells of the vasculature.
In the long term, ideally, physiological relevance would be demonstrated. It would be useful to investigate if shuttling of full length TF mRNA results in significantly increased thrombogenicity of recipient cells, or if CXCL10 mRNA shuttle is appreciably pro-inflammatory. Perhaps, for example, mRNA shuttle could be used to attempt to temporarily restore thrombogenicity to TF-knockout mouse cells. A potential complication would be that as TF protein is also shuttled via microparticles, one would have to devise a method to differentiate between the effects of transferred protein and transferred mRNA. It might be possible to grow recipient cells in media tagged with a heavy isotope such as Carbon 13 or Nitrogen 15 and hence differentiate between protein synthesized “on site” and protein transferred from donor cells, which would incorporate the common isotope. Intriguingly, this would also allow the synthetic pathways of the two proteins to be followed separately. It would be very interesting if the processing pathways were different or if the end products varied structurally.
This brings us back to the idea of what RBPs are associated with the shuttled mRNA, and the potential biological implications. The model that was sketched in the Introduction as a basis for experimenting with TF and CXCL10 mRNA would suggest that these transcripts might be complexed with PARP14 and/or HuR, which may form part of an RNP complex whose constituents represent a code to determine how the transcript is processed as well as to possibly modulate other gene expression in the recipient cell. In this example, the accompanying RNP complex might, hypothetically, encourage the synthesis of a TF isoform that is more easily decrypted [7]. TF decryption is a transition whereby TF switched from a low-activity to a fully active form. TF decryption is generally associated with loss of membrane phospolipid asymmetry as well as (speculatively) with a transition from dimeric to monomeric TF7. Modulation of post-translational modifications in TF, which may be earmarked at the transcript stage, could encourage active conformations over inactive ones. Alternatively, RNP modulation of the expression of other genes could achieve a similar effect: that of a relevant glycosylase or chaperone protein for example. The regulatory possibilities inherent in the intercellular transfer of gene expression-modulating RNP complexes are legion.
In conclusion, I have some evidence that the 3’UTRs of TF and CXCL10 mRNA may direct intercellular shuttling, possibly via a microparticle vehicle. If true, it would be of interest to discover the nature, if any, of any RNP complexes to which the shuttling mRNA belongs. Such a phenomenon provides a mechanism by which thrombogenic and inflammatory responses may be coordinated between cells at the transcriptional level by RNA- and RNP-centric regulation. Further insight into such a mechanism would have therapeutic and diagnostic applications in many pathologies that involve prolonged thrombotic risk and/or inflammation, such as coronary artery disease, which has an accompanying risk of pulmonary artery thrombosis. More generally, the intercellular transfer of RNA and possibly of gene expression-modulating RNP complexes represents an expansion of regulatory complexity that invites exploration.
MATERIALS AND METHODS
PCR and RT-PCR
All PCR reactions utilized 200 M dNTPs, 10 M of the forward and reverse primers each, variable quantities of template DNA or cDNA, 1x AD2 PCR buffer and 1x AD2 polymerase (Clontech Advantage® 2 PCR Kit). All PCR reactions utilized the following thermocycler program: 1 min at 94oC, (30 sec at 94oC, 30 sec at 55oC, 30 sec at 72oC) x 42, and 5 min at 4oC.
The following PCR primers (Eurofins) were used in this project:
TF 3’UTR forward - AGGAAAGGCTGAAGCCG
TF 3’UTR reverse - GCGATTGTCTGAATTCC
CXCL10 3’UTR forward - CTCCTTAACTGGAGTGAAGC
CXCL10 3’UTR reverse - TTTCATGTCAGTTATTATTACTTTATTAC
GFP forward - CGACACAATCTGCCCTTTCG
BGH reverse - TAGAAGGCACAGTCGAGG
All RT-PCR reactions utilized SuperScript III Reverse Transcriptase (Invitrogen) and the associated protocol, with the following modifications: for a 20 l reaction, the initial (pre-65 oC) 13 l reaction included 11 l of purified RNA solution and no sterile water. The RNaseOUT Recombinant RNase Inhibitor was replaced with sterile water. RT-PCR was specific for construct mRNA by using the GFP forward primer and a 3’UTR reverse primer specific to either TF or CXCL10.
Agarose gel electrophoresis
DNA length and purity were verified by loading 10 l samples of PCR reaction with 1x gel loading buffer (Sigma) into wells of a 1% w/v agarose gel spiked with 0.6 g/ml ethidium bromide. The gels were run at 80V for an appropriate time. DNA was visualized with a transilluminator.
Construct engineering
813 bp of the TF 3’UTR was PCR amplified from full-length template DNA. 729 bp of the CXCL10 3’UTR was PCR amplified from full-length template DNA. PCR protocol was as described above with an additional 1 min step at 72oC prior to final cooling. Gel electrophoresis was used to verify 3’UTR DNA length and purity as above. DNA was purified from the gel using QIAquick Gel Extraction Kit (Qiagen) as per the associated protocol. The 3’UTR sequences were then cloned into pcDNA3.1/NT-GFP-TOPO plasmid vectors, which were subsequently transformed into One Shot TOP10 Chemically Competent Cells using GFP Fusion TOPO® TA Expression Kits (Invitrogen) as per the associated protocol. Transformed cells were plated on LB agar (Sigma) plates and incubated for 16 hrs at 37oC. 8 single colonies for each construct were used to separately inoculate 5ml aliquots of LB broth (Sigma) and incubated for 16 hrs at 37oC. DNA was purified using QIAprep Miniprep kits (Qiagen) as per the associated protocol. Purified DNA from the 8 clones of each construct was sent for sequencing. One clone for each construct with complete fidelity to template sequences was chosen and used to inoculate 200ml of LB broth, which was incubated for 16 hrs at 37oC. Endotoxin-free construct DNA was purified using QIAprep Maxiprep kits (Qiagen) and used for transfection.
Cell culture and transfection
HeLa cells were cultured in DMEM with 10% FCS and 2 mM L-glutamine at 37oC in 5% CO2. Before each transfection or supernatant transfer event, Hela cells were passaged and seeded at a density of 8 x 104 cells/ml in 6 well plates 24 hours before the event. All transfection was transient and produced using 2 g/well of plasmid DNA and 5 l/well of lipofectamine 2000 reagent (Invitrogen) diluted in Opti-MEM Reduced Serum Medium (Invitrogen) as per the associated protocol.
LPS stimulation was achieved using a final concentration of 1 g/ml. IFN stimulation was achieved using a final concentration of 2.5 ng/ml. For the UV-irradiated control, HeLa cells were transfected as normal. 4 hours after transfection, cells were washed with PBS and given fresh medium. Subsequently, cells were irradiated in their wells with 312 nm UV light at 180 W for 10 min from a Uvitec Transilluminator STX-40.M. Cells were replaced in incubation for the remaining 12 hours. Prior to culture medium collection, their morphology was checked by light microscopy.
Supernatant transfer and treatments
Culture medium from donor HeLa cells was collected and centrifuged at 1000 g for 5 min to pellet cells and debris. Culture supernatant was treated or left untreated, then used to replace culture medium of untransfected recipient HeLa cells.
Supernatant treatments are outlined below:
DNase: 1ml aliquot of culture supernatant was treated with DNA-free kit (invitrogen) as per the associated protocol. 2 l of DNase I were used per aliquot.
RNase: culture supernatant was treated with 100 g/ml of RNase A (Qiagen) and incubated for 60 min at 37oC.
Protease: culture supernatant was treated with 1 g/ml of Proteinase K (Qiagen) and incubated for 60 min at 37oC.
DNase I control was performed by spiking 1 ml of sterile culture medium (DMEM with 10% FCS and 2 mM L-glutamine) with 1 g of GFP/TF-3’UTR plasmid DNA and vortexing for 30 sec. The sterile medium was then treated with DNA-free kit (invitrogen) as per the associated protocol. 2 l of DNase I were used.
Viable cell count was performed washing adherent cells twice with PBS, then trypsinising them for 5 min. Trypsin was inactivated by adding culture medium. A 20 l aliquot was removed and added to 20 l of Trypan blue. Viable (non-blue) cell concentration was determined thrice with a haemocytometer and light microscrope. The average of the three readings was taken.
Western Blot
Western blot was performed as previously described10 with the following modifications: after boiling, sample were loaded into a NuPAGE® 4-12% Bis-Tris Gel (1.5 mm, 10 well) and run for 45 min at 180 V in MES buffer. Blotting onto nitrocellulose membrane was performed using Tris-Glycine transfer buffer (20% v/v methanol) at 25 V for 120 min. After transfer, membrane was blocked with 5% marvel (milk powder) for 60 min, then incubated for 16 hours in 5% marvel and 1:200 mouse anti-GFP (primary). Membrane was washed in PBS thrice and incubated for 1 hour with 5% marvel and 1:2000 rabbit anti-mouse (secondary, horseradish-peroxidase conjugated). Membrane was washed a further three time in PBS. Each PBS wash was 10 min. ECL plus was used to visualize antibody distribution as per associated protocol.
RNA purification
RNA was purified from biological pellets using RNeasy Minikit (Qiagen) as per the associated protocol. RNA was purified from biological fluids using Trizol LS Reagent (Ambion) as per the associated protocol. RNA purification included a DNase treatment step.
Confocal microscopy
For mounting, HeLa cells (grown on discs) were washed thrice in PBS, the cells left unstained or their nuclei stained with TOPRO (Invitrogen) diluted 1:1000 in BSA and washed twice again. Cells were then fixed in 5% paraformaldehyde for 15 min then washed twice in PBS. Discs were mounted cells-down on coverslips using VECTASHIELD Mounting Medium (VectorLabs) and sealed with nail varnish. Images were captured using an LSM510 META confocal microscope (Carl Zeiss) on the 40x objective lens, and running Version 3.2 of the LSM acquisition software.
Differential centrifugation
Low-speed centrifugation was performed at 5000 g for 10 min, 10 000 g for 10 min, 18 000 g for 30 min and 22 000 g for 30 min. High-speed centrifugation was performed at 5000 g for 10 min, 22 000 g for 10 min, and 150 000 g for 60 min. Centrifugation at or below 22 000 g was achieved with a standard bench-top centrifuge. Centrifugation above 22 000 g was achieved with a SW 40 Ti rotor (Beckman optima LE-80k Ultracentrifuge). All centrifugation was performed at 4oC.
REFERENCES
1. Keene (2007) “RNA regulons: coordination of
post-transcriptional events” Nature 8:533
2. Mansfield at al. (2009) “The ribonome: a dominant force in
co-ordinating gene expression” Biol. Cell 101:169
3. Valadi et al. (2007) “Exosome-mediated transfer of mRNAs and microRNAs
is a novel mechanism of genetic exchange between cells” Nature 9:654
4. Orozco et al. (2010) “Flow Cytometric Analysis of Circulating Microparticles
in Plasma” Cytometry 77A: 502
5. Camussi et al. (2010) “Exosomes/microvesicles as a mechanism of
cell-to-cell communication” Kidney International 78:838
6. Unpublished work (2012). Haskard laboratory, National Heart & Lung Institute, London. Address correspondence to d.haskard@imperial.ac.uk
7. Nigel et al. (2010) “Tissue Factor and Its Measurement in Whole
Blood, Plasma, and Microparticles” Seminars in Thrombosis and Hemostasis 36:865
8. Aharon et al. (2008) “Monocyte-derived microparticles and exosomes induce
procoagulant and apoptotic effects on endothelial cells” Thromb Haemost 100: 878
9. Ardoin et al. (2007) “The Role of Microparticles in Inflammation and
Thrombosis” Scandinavian Journal of Immunology 66:159
10. Stoorvogel et al. (1989) “Relations between the intracellular pathways of the receptors for transferrin, asialoglycoprotein, and mannose 6-phosphate in human hepatoma cells” J. Cell Biol. 108:2137
11. Personal communication (2012). Michael Johns, Research Associate at the National Heart & Lung Institute, London. Address correspondence to m.johns@imperial.ac.uk
12. Ostrowski et al. (2009) “Rab27a and Rab27b control different steps of the
exosome secretion pathway” Nature 12:19
13. Secchini et al. (2005) “Interstrand cross-link induction by UV radiation in bromodeoxyuridine-substituted DNA: dependence on DNA conformation” Biochemistry 44:16957
14. Koditz et al. (2002) “Dissecting the effect of trifluoroethanol on ribonuclease A: Subtle structural changes detected by nonspecific proteases” Eur. J. Biochem. 269:3831
15. Donny et al. (2010) “RNA processing and its regulation: global insights into biological networks” Nature 11:75
16. Park et al. (2006). "Human umbilical vein endothelial cells and human dermal microvascular endothelial cells offer new insights into the relationship between lipid metabolism and angiogenesis". Stem Cell Rev 2:93
コメント